Riney K, Bogacz A, Somerville E, Hirsch E, Nabbout R, Scheffer IE, et al. International League Against Epilepsy classification and definition of epilepsy syndromes
with onset at a variable age: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63:1443–1474.
https://doi.org/10.1111/epi.17240
Lafora Disease (LD) also known as Lafora Body Disease is a progressive myoclonic epilepsy that has an age of onset between 6–19 years of age. Early rapid cognitive decline, vision, and motor deterioration, cerebellar symptoms (ataxia, incoordination) are the clinical manifestations typically presented. This disease is fatal approximately a decade after its onset.
Patients can have focal seizures with visual symptoms as an early feature and with progression myoclonic and generalized tonic–clonic seizures. Myoclonic seizures gradually worsen and become intractable, and progressive cognitive decline continues. By 10 years after onset, affected individuals have nearly continuous myoclonus with absence seizures, frequent generalized tonic–clonic seizures, and profound dementia or are in a vegetative state.
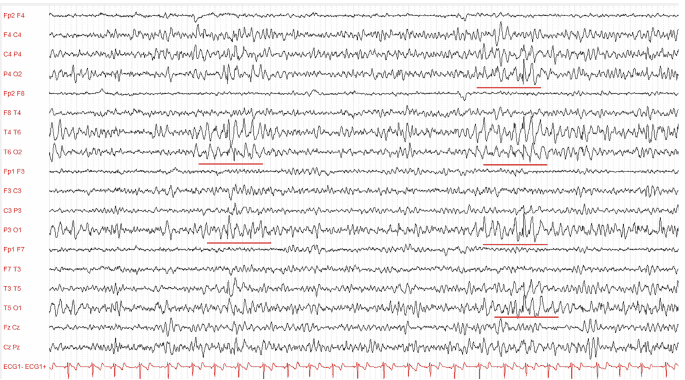
At onset, the EEG has a normal background, with interictal spike-and-wave and polyspike discharges that are activated by photic stimulation at low frequencies. With time, the EEG background slows, and epileptiform abnormality increases in frequency and may have emphasis in posterior regions. Patients with Lafora disease can develop erratic myoclonus without EEG correlation.
Question:
Which of the following diagnostic or imaging tests can show the metabolism of the glucose in the brain that can aid in determining the progression of Lafora Disease?
Results
#1. Which of the following diagnostic or imaging tests can show the metabolism of the glucose in the brain that can aid in determining the progression of Lafora Disease?
Fluorodeoxyglucose positron emission tomography (PET) can show extensive areas of decreased glucose metabolism, the severity of which may correlate with stage of disease. Pathogenic gene variants in EPM2A (laforin) and EPM2B (malin) are found in 70% and 27% of cases, respectively, with no pathogenic variant found in 3%. Lafora bodies (accumulation of glycogen) are seen in sweat duct cells and in other tissues. This condition is differentiated from ULD by the presence of early cognitive decline and rapid progression of the PME.
MRI is usually normal, but magnetic resonance spectroscopy may show significant reduction of the N-acetylaspartate/creatine ratio in frontal cortex, basal ganglia, and cerebellar hemispheres.



